E. Computation of Polarizable Molecular Block Model Using
Non-bonded Induced Dipole Force Field
Here, the energy calculations of trinucleotides and trinucleotide duplex are performed using polarizable molecular block model.
The Fortran program IDFFpmb_mi is used for the energy evaluation.
This program is just calculate energies, dipole moments, electrostaioc potentials and molecular polarizabilities of target molecules.
Reproducibilities of high-level QM (wB97XD/aug-cc-pVDZ) results are examined.
The calculation and analysis are performed in the following steps:
1. Reference QM calculations of trinucleotide duplexe
'runG09P_rest_scf.cm' is the csh command to run G09P calculations. Five jobs of trinucleotide duplex and trinucleotides are submitted. Here the BSSE-corrected interaction energies are calculated. The ESP of the molecular surface is output (Rest_scf). The file name is changed to wB97XDaD_GACGTC_028.8_scf.rest in the case of trinucleotide duplex.
Download GACGTC
'wB97XDaD_GAC.gjf' is the G09 input file for trinucleotide (sGAC). The route section is as follows:
Download GAC GTC GACGTC
GACghost ghostGTC
'wB97XDaD_GAC.out' is the G09P output of sGAC.
Download GAC GTC GACGTC
'wB97XDaD_GAC.rest' contains atomic coordinates, atomic charges, molecular surface ESP and surface coordinates of sGAC. This file is used as input data for the polarizable molecular block model.
Download GAC GTC GACGTC
- Reference QM calculations of trinucleotide duplexes
- Calculation of trinucleotide duplexe using dumping type S3
- Calculation of trinucleotide duplexe using dumping type G
- Calculation results of trinucleotide duplexe using polarizable molecular block model
1. Reference QM calculations of trinucleotide duplexe
'runG09P_rest_scf.cm' is the csh command to run G09P calculations. Five jobs of trinucleotide duplex and trinucleotides are submitted. Here the BSSE-corrected interaction energies are calculated. The ESP of the molecular surface is output (Rest_scf). The file name is changed to wB97XDaD_GACGTC_028.8_scf.rest in the case of trinucleotide duplex.
1) Trinucleotide duplex job (GACGTC)
2) Trinucleotide jobs (GAC and GTC)
3) Trinucleotide jobs with ghost orbitals jobs (GACghost and ghostGTC)
2) Trinucleotide jobs (GAC and GTC)
3) Trinucleotide jobs with ghost orbitals jobs (GACghost and ghostGTC)
Download GACGTC
'wB97XDaD_GAC.gjf' is the G09 input file for trinucleotide (sGAC). The route section is as follows:
1) Calculation level (DFT/basis set) is wB97XD/aug-cc-pVDZ.
2) The "pop=MK" and "iop(6/20=8)" option output ESP with vdw1.8 times kosugi point selection. (Required)
3) The iop(2/11=1) option prints the "input orientation" coordinates.
4) The "scf=XQC" option is useful in cases of difficult convergence.
5) "Int=(ultrafine,Acc2E=12)" is an option for numerical integration. The default is FineGrid and 10.
6) The "polar" option is for the calculation of molecular polarizability.
Input files for the trinucleotides (GACGTC and GTC) are also available for download.
Input files for trinucleotides with ghost orbitals (GACghost and ghostGTC) are also available for download. "Massage" option has been added. 2) The "pop=MK" and "iop(6/20=8)" option output ESP with vdw1.8 times kosugi point selection. (Required)
3) The iop(2/11=1) option prints the "input orientation" coordinates.
4) The "scf=XQC" option is useful in cases of difficult convergence.
5) "Int=(ultrafine,Acc2E=12)" is an option for numerical integration. The default is FineGrid and 10.
6) The "polar" option is for the calculation of molecular polarizability.
Download GAC GTC GACGTC
GACghost ghostGTC
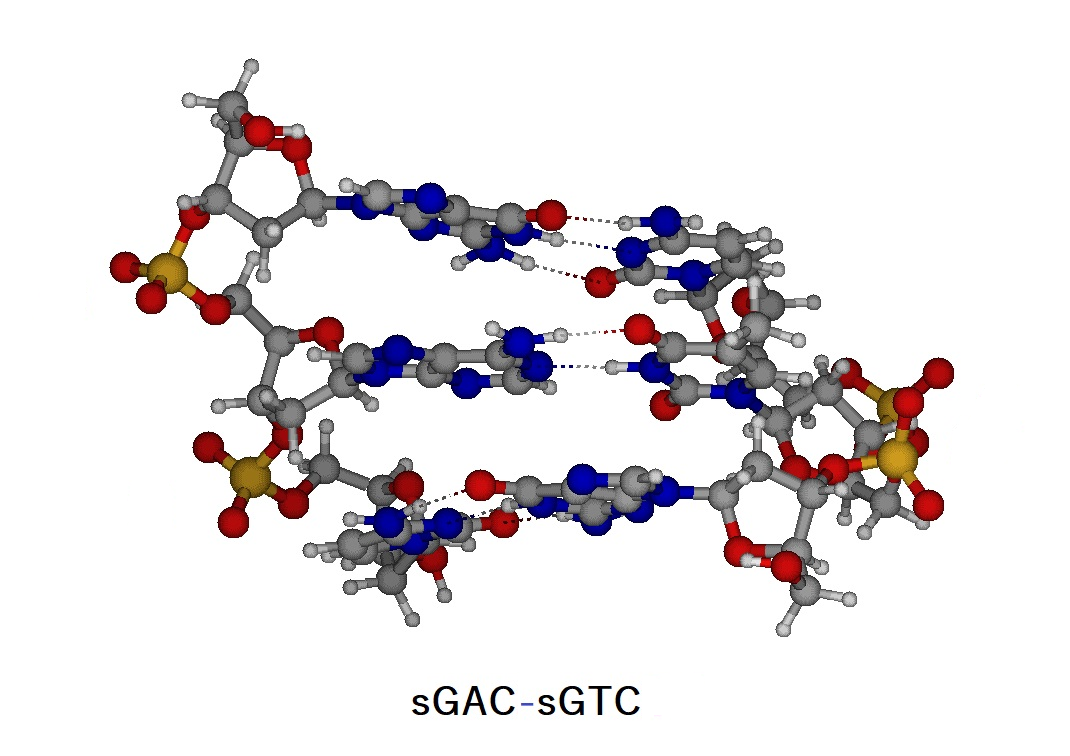
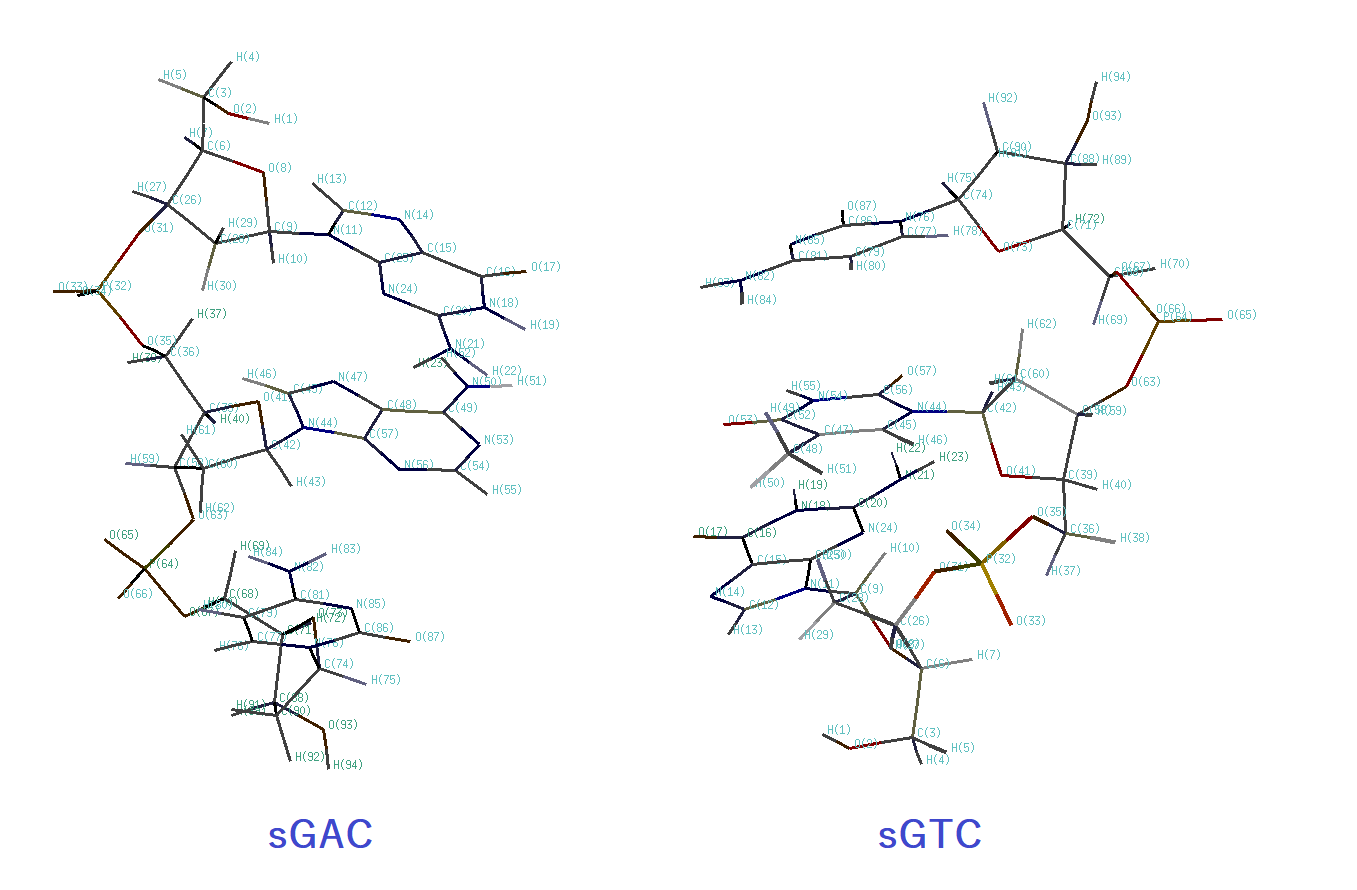
Figure E1. Structure of the trinucleotide duplex (sGAC-sGTC) and atomic numbering of the trinucleotides (sGAC and sGTC).
'wB97XDaD_GAC.out' is the G09P output of sGAC.
Download GAC GTC GACGTC
'wB97XDaD_GAC.rest' contains atomic coordinates, atomic charges, molecular surface ESP and surface coordinates of sGAC. This file is used as input data for the polarizable molecular block model.
Download GAC GTC GACGTC
2. Calculation of trinucleotide duplexe using dumping type S3
'idffpmb_mi.cm' is a sample csh command to run the PMB model using the non-bonded induced dipole FF. The dumping type is S3. GAC-GTC, GAC, and GTC jobs are submitted.
Download
'top_r13_smallGACsGTCs_wB_aT_cT_s03111_one.inp' is the TopPar file for GAC and GTC. 'top_r13_smallGACsGTCs_wB_aT_cT_s03111_one.inp' is prepared in 'IDFF TopPar' page. 'GACs' and 'GTCs' are the residue name.
Download
'seq_GACsGTCs.inp' specifies a sequence.
Download
'scp_100.dat' specifies the coefficient to uniformly multiply the atomic polarizability. Here we specify "1.0" which does nothing.
Download
'100idff_GAC_s03111_one.out' is the output file for GAC. GACGTC and GTC output files are also available for download.
Download
'IDFFpmb_mi_2022.f' is a FORTRAN program for polalizable molecular block model using non-bonded induced dipole force field. If you downloaded and used the IDFFpmb program, please inform S. Nakagawa that you used it (naka(a)kinjo-u.ac.jp). Also, let me know if you find any bugs.
Download
'idffpmb_mi.cm' is a sample csh command to run the PMB model using the non-bonded induced dipole FF. The dumping type is S3. GAC-GTC, GAC, and GTC jobs are submitted.
1) 'top_r13_smallGACsGTCs_wB_aT_cT_s03111_one.inp' is a topology-parameter (TopPar) file for GAC and GTC.
2) 'seq_GACsGTCs.inp' is the input file for the sequence. (fort.2)
3) 'wB97XDaD_GACGTC_028.8_scf.rest' is the ESP file above. (fort.3)
4) 'scp_100.dat' is the input file for atomic polarizability. The scale factor data is ' 1.0' (unscaled). (fort.4)
5) 'IDFFpmb_mi.exe' is the executable file of the PMB model.
6) '100idff_GACGTC_028.8_s03111_one.out' is the output file. (fort.10)
2) 'seq_GACsGTCs.inp' is the input file for the sequence. (fort.2)
3) 'wB97XDaD_GACGTC_028.8_scf.rest' is the ESP file above. (fort.3)
4) 'scp_100.dat' is the input file for atomic polarizability. The scale factor data is ' 1.0' (unscaled). (fort.4)
5) 'IDFFpmb_mi.exe' is the executable file of the PMB model.
6) '100idff_GACGTC_028.8_s03111_one.out' is the output file. (fort.10)
Download
'top_r13_smallGACsGTCs_wB_aT_cT_s03111_one.inp' is the TopPar file for GAC and GTC. 'top_r13_smallGACsGTCs_wB_aT_cT_s03111_one.inp' is prepared in 'IDFF TopPar' page. 'GACs' and 'GTCs' are the residue name.
Download
'seq_GACsGTCs.inp' specifies a sequence.
Download
'scp_100.dat' specifies the coefficient to uniformly multiply the atomic polarizability. Here we specify "1.0" which does nothing.
Download
'100idff_GAC_s03111_one.out' is the output file for GAC. GACGTC and GTC output files are also available for download.
Download
'IDFFpmb_mi_2022.f' is a FORTRAN program for polalizable molecular block model using non-bonded induced dipole force field. If you downloaded and used the IDFFpmb program, please inform S. Nakagawa that you used it (naka(a)kinjo-u.ac.jp). Also, let me know if you find any bugs.
Download
3. Calculation of trinucleotide duplexe using dumping type G
'idffpmb_mi_g.cm' is a sample csh command to run the PMB model using the non-bonded induced dipole FF. The dumping type is G. GAC-GTC, GAC, and GTC jobs are submitted.
Download
'top_r163_smallGACsGTCs_wB_aT_cT_g0926_one.inp' is the TopPar file for GAC and GTC. 'top_r163_smallGACsGTCs_wB_aT_cT_g0926_one.inp' is prepared in 'IDFF TopPar' page. 'GACs' and 'GTCs' are the residue name.
Download
'100idff_GAC_g0926_one.out' is the output file for GAC. GACGTC and GTC output files are also available for download.
Download
'idffpmb_mi_g.cm' is a sample csh command to run the PMB model using the non-bonded induced dipole FF. The dumping type is G. GAC-GTC, GAC, and GTC jobs are submitted.
1) 'top_r13_GACstGTCst_wB_aT_cT_g0926_one.inp' is a topology-parameter (TopPar) file for GAC and GTC.
2) 'seq_GACsGTCs.inp' is the input file for the sequence. (fort.2)
3) 'wB97XDaD_GACGTC_028.8_scf.rest' is the ESP file above. (fort.3)
4) 'scp_100.dat' is the input file for atomic polarizability. The scale factor data is ' 1.0' (unscaled). (fort.4)
5) 'IDFFpmb_mi.exe' is the executable file of the PMB model.
6) '100idff_GACGTC_028.8_g0926_one.out' is the output file. (fort.10)
2) 'seq_GACsGTCs.inp' is the input file for the sequence. (fort.2)
3) 'wB97XDaD_GACGTC_028.8_scf.rest' is the ESP file above. (fort.3)
4) 'scp_100.dat' is the input file for atomic polarizability. The scale factor data is ' 1.0' (unscaled). (fort.4)
5) 'IDFFpmb_mi.exe' is the executable file of the PMB model.
6) '100idff_GACGTC_028.8_g0926_one.out' is the output file. (fort.10)
Download
'top_r163_smallGACsGTCs_wB_aT_cT_g0926_one.inp' is the TopPar file for GAC and GTC. 'top_r163_smallGACsGTCs_wB_aT_cT_g0926_one.inp' is prepared in 'IDFF TopPar' page. 'GACs' and 'GTCs' are the residue name.
Download
'100idff_GAC_g0926_one.out' is the output file for GAC. GACGTC and GTC output files are also available for download.
Download
4. Calculation results of trinucleotide duplexe using polarizable molecular block model
The PMB results of monomer trinucleotides (sGAC and sGTC) are shown in Table E1. The referenced QM calculation is wB97XD/aug-cc-pVDZ. The root mean square deviation of ESP is 2 kcal/mol for dumping types S3 and G. On the other hand, it is 7-8 kcal/mol for block charges only. ESP on the molecular surface has been better recapitulated by the introduction of PMB. The coordinate dependent dipole moments are reproduced well. The reproducibility of x, y, and z components are also well. QM molecular polarizabilities are calculated using wB97XD/aug-cc-pVDZ and wB97XD/cc-pVTZ (shown in the parentethes). Since the atomic polarizabilities of block molecules are optimized based on the B97XD/cc-pVTZ calculations, the molecular polarizabilities calculated by wB97XD/cc-pVTZ are well reproduced by PMB. The reproducibility of xx, yy, and zz components are also well.
a ESP relative rms deviation is shown in parenthesis.
b Unit of polarizability is Bohr3.
c Molecular polarizability of wB97XD/cc-pVTZ is shown in parenthesis.
The PMB results of trinucleotide duplex (sGAC-sGTC) are shown in Table E2. The referenced QM calculation is wB97XD/aug-cc-pVDZ. The root mean square deviation of ESP is 2-3 kcal/mol for dumping types S3 and G. On the other hand, it is 6 kcal/mol for block charges only. ESP on the molecular surface has been better recapitulated by the introduction of PMB. The coordinate dependent dipole moments are reproduced well. The reproducibility of x, y, and z components are also well. QM molecular polarizabilitie is calculated using wB97XD/cc-pVTZ (shown in the parentethes). The molecular polarizability calculated by wB97XD/cc-pVTZ is well reproduced by PMB. The reproducibility of xx, yy, and zz components are also well.
a Interaction energy of ΔEes+ΔEvdw is shown.
b ESP relative rms deviation is shown in parenthesis.
c Unit of polarizability is Bohr3.
d Molecular polarizability of wB97XD/cc-pVTZ is shown in parenthesis.
The PMB results of monomer trinucleotides (sGAC and sGTC) are shown in Table E1. The referenced QM calculation is wB97XD/aug-cc-pVDZ. The root mean square deviation of ESP is 2 kcal/mol for dumping types S3 and G. On the other hand, it is 7-8 kcal/mol for block charges only. ESP on the molecular surface has been better recapitulated by the introduction of PMB. The coordinate dependent dipole moments are reproduced well. The reproducibility of x, y, and z components are also well. QM molecular polarizabilities are calculated using wB97XD/aug-cc-pVDZ and wB97XD/cc-pVTZ (shown in the parentethes). Since the atomic polarizabilities of block molecules are optimized based on the B97XD/cc-pVTZ calculations, the molecular polarizabilities calculated by wB97XD/cc-pVTZ are well reproduced by PMB. The reproducibility of xx, yy, and zz components are also well.
| wB97XD/ aug-cc-pVDZ (cc-pVTZ) | PMB S3 | PMB G 0.926 | block charge | |
|---|---|---|---|---|
| sGAC ESP rmsd (kcal/mol) | 0.0 | 2.18 (2.37%)a | 2.43 (2.64%) | 7.34 (7.99%) |
| sGTC ESP rmsd (kca/mol) | 0.0 | 2.18 (2.38%)a | 2.36 (2.58%) | 7.53 (8.25%) |
| sGAC Dipole Moment μ (D) | 17.33 | 16.69 | 16.09 | 25.08 |
| μx | 0.51 | 1.42 | 1.79 | 0.43 |
| μy | 16.92 | 16.24 | 15.62 | 24.98 |
| μz | -3.73 | -3.57 | -3.44 | -2.17 |
| sGTC Dipole Moment μ (D) | 15.62 | 15.23 | 14.69 | 23.93 |
| μx | 0.87 | -0.31 | -0.63 | 1.39 |
| μy | -15.59 | -15.22 | -14.67 | -23.84 |
| μz | -0.63 | -0.30 | -0.23 | 1.48 |
| sGAC Molecular Polarizabilityb αisotropic | 517.5 (481.6) c | 479.1 | 481.4 | - |
| αxx | 545.3 (511.1) | 509.9 | 514.5 | - |
| αyy | 575.0 (537.6) | 539.2 | 542.9 | - |
| αzz | 432.1 (396.2) | 388.1 | 386.8 | - |
| sGTC Molecular Polarizabilityb αisotropic | 505.2 (470.1) c | 466.9 | 467.7 | - |
| αxx | 528.9 (493.9) | 493.6 | 493.8T | - |
| αyy | 538.7 (502.3) | 501.3 | 505.1 | - |
| αzz | 447.9 (414.2) | 405.8 | 404.1 | - |
The PMB results of trinucleotide duplex (sGAC-sGTC) are shown in Table E2. The referenced QM calculation is wB97XD/aug-cc-pVDZ. The root mean square deviation of ESP is 2-3 kcal/mol for dumping types S3 and G. On the other hand, it is 6 kcal/mol for block charges only. ESP on the molecular surface has been better recapitulated by the introduction of PMB. The coordinate dependent dipole moments are reproduced well. The reproducibility of x, y, and z components are also well. QM molecular polarizabilitie is calculated using wB97XD/cc-pVTZ (shown in the parentethes). The molecular polarizability calculated by wB97XD/cc-pVTZ is well reproduced by PMB. The reproducibility of xx, yy, and zz components are also well.
| wB97XD/ aug-cc-pVDZ (cc-pVTZ) | PMB S3 | PMB G 0.926 | block charge | |
|---|---|---|---|---|
| ΔE (kcal/mol) | 19.8 | 18.1 | 15.9 | 30.5a |
| ESP rmsd (kcal/mol) | 0.0 | 2.46 (1.74%)b | 2.71 (1.91%) | 5.96 (4.20%) |
| Dipole Moment μ (D) | 4.36 | 4.48 | 4.40 | 9.27 |
| μx | 0.74 | 0.44 | 0.32 | 1.14 |
| μy | -2.35 | -0.87 | -0.48 | -8.58 |
| μz | 3.59 | 4.37 | 4.36 | 3.31 | Molecular Polarizabilityc αisotropic | (976.1) d | 966.2 | 970.5 | - |
| αxx | (1192.1) | 1183.3 | 1195.3 | - |
| αyy | (988.9) | 985.7 | 990.1 | - |
| αzz | (747.2) | 729.6 | 726.2 | - |