C. Electrostatic Potential Fitting
The restart procedure for ESP optimization is described. The calculation is performed in the following steps:
1. Adjust the restart file with respect to atoms and ESP before ESP fitting
Specifying the "pop=MK iop(6/20=8)" option in G09 gives the atomic charges fitted with ESP. However, in the PMB model, the capped hydrogens between block molecules are duplicated, so we need to remove them and reoptimize to fill the integer charges. The PMB model also removes molecular surface points that are within 2.2 Å of the capped hydrogen.
"redaddxyz.cm" is the csh command to adjust atom and ESP before reoptimization.
Download
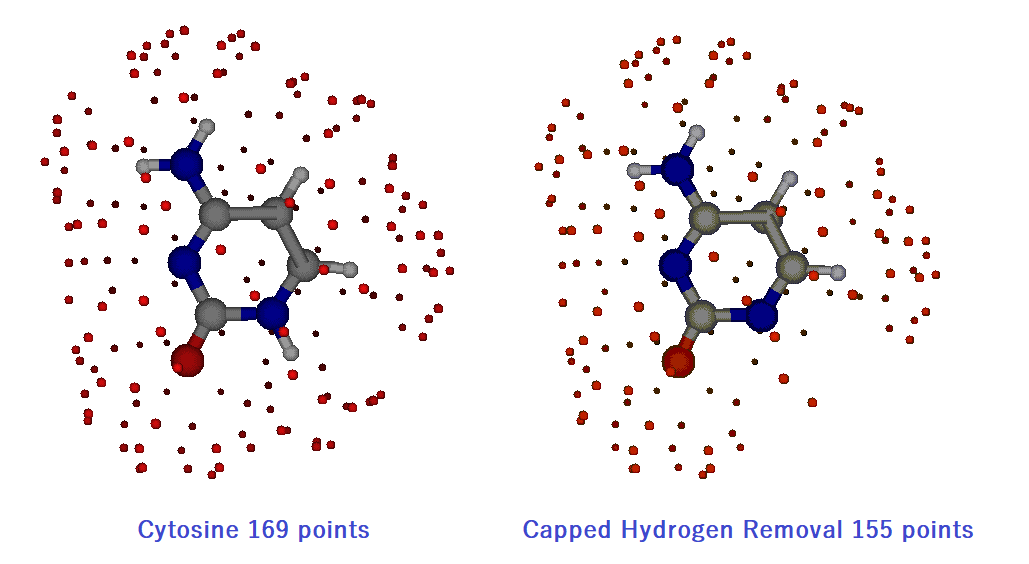
The restart data of ESP is "opt_scf_C_aT.rest" (see "G09 Mod ESP" ). This cytosine data has 13 atoms and 337 surface selection points. If the number of surface points is greater than 300, reduce it by half. Here it is 337 points, so it is reduced to 169 points as shown in above figure.
"red1.dat" specifies the atom to remove. Here the first atom is removed. You can remove up to 3 atoms. Points within the 2.2 Å from the removed H atom are also erased. Here, 14 points are erased, so the surface points are 155 as shown above.
Download
The output "red1.rest" is used as the restart file for ESP refitting. This data has 12 atoms and 155 surface selection points. If the total charge is not 0.0 e, manually change the value in line 1 of red1_rest file. Atomic charges optimized by G09 are entered in the rightmost columns of rows 2-13. These will be the initial values during reoptimization. In molecular mechanics (MM), the charges of the H atoms of -CH3 and -NH2 groups are equally assigned. In red1.rest same value (0.47) is assigned for the amino group hydrogens of cytosine. Reoptimization can treat the same value as one variable, so manually change it to the same value before optimization.
Download
"redaddxyz.f" is a Fortran program.
Download
- Adjust the restart file with respect to atoms and ESP before ESP fitting
- ESP optimization to determine point charges
- Impact of Capped Hydrogen Removal Operation
1. Adjust the restart file with respect to atoms and ESP before ESP fitting
Specifying the "pop=MK iop(6/20=8)" option in G09 gives the atomic charges fitted with ESP. However, in the PMB model, the capped hydrogens between block molecules are duplicated, so we need to remove them and reoptimize to fill the integer charges. The PMB model also removes molecular surface points that are within 2.2 Å of the capped hydrogen.
"redaddxyz.cm" is the csh command to adjust atom and ESP before reoptimization.
Download
The restart data of ESP is "opt_scf_C_aT.rest" (see "G09 Mod ESP" ). This cytosine data has 13 atoms and 337 surface selection points. If the number of surface points is greater than 300, reduce it by half. Here it is 337 points, so it is reduced to 169 points as shown in above figure.
"red1.dat" specifies the atom to remove. Here the first atom is removed. You can remove up to 3 atoms. Points within the 2.2 Å from the removed H atom are also erased. Here, 14 points are erased, so the surface points are 155 as shown above.
Download
The output "red1.rest" is used as the restart file for ESP refitting. This data has 12 atoms and 155 surface selection points. If the total charge is not 0.0 e, manually change the value in line 1 of red1_rest file. Atomic charges optimized by G09 are entered in the rightmost columns of rows 2-13. These will be the initial values during reoptimization. In molecular mechanics (MM), the charges of the H atoms of -CH3 and -NH2 groups are equally assigned. In red1.rest same value (0.47) is assigned for the amino group hydrogens of cytosine. Reoptimization can treat the same value as one variable, so manually change it to the same value before optimization.
Download
"redaddxyz.f" is a Fortran program.
Download
2. ESP optimization to determine point charges
"espopt_red1.cm" is a sample csh command to run ESP reoptimization. "red1.rest" is a input ESP file. "irest.dat" specifies "1".
Download
"red1.out" is a output file. The optimized charges are obtained.
Download
"fin_chg_C_red1.dat" is the optimized point charge extraction file.
Download
"ESPopt.f" is a Fortran program for ESP-driven charge repotimization. "ESPopt.exe" is the executable file.
A nonlinear optimization (Levenberg-Marquardt) procedure is used. The original creator of this program is unknown, but it was already used in Karplus's laboratory in 1985. This program is now used to re-optimize the ESP charges. The original program has been extensively modified to allow optimization of atomic polarizabilities as well, but this function has been taken over by a separate, independent program, POPopt, so the ESPopt is not used for polarizability optimization purposes. Note that the POP optimization results are not guaranteed. The ESP charge re-optimization results are available.
Download
"espopt_red1.cm" is a sample csh command to run ESP reoptimization. "red1.rest" is a input ESP file. "irest.dat" specifies "1".
Download
"red1.out" is a output file. The optimized charges are obtained.
Download
"fin_chg_C_red1.dat" is the optimized point charge extraction file.
Download
"ESPopt.f" is a Fortran program for ESP-driven charge repotimization. "ESPopt.exe" is the executable file.
A nonlinear optimization (Levenberg-Marquardt) procedure is used. The original creator of this program is unknown, but it was already used in Karplus's laboratory in 1985. This program is now used to re-optimize the ESP charges. The original program has been extensively modified to allow optimization of atomic polarizabilities as well, but this function has been taken over by a separate, independent program, POPopt, so the ESPopt is not used for polarizability optimization purposes. Note that the POP optimization results are not guaranteed. The ESP charge re-optimization results are available.
Download
3. Impact of Capped Hydrogen Removal Operation
Even when the number of points on the cytosine surface is reduced by half (169 points), the rms deviation of ESP tends to be almost the same as shown in the table below. Here the charges of the two hydrogens (H41 and H42) of the amino group are optimized to the same value. The molecular dipole moment is the same as the 337 points optimization. In the case of 155 points where capped hydrogen removal (CHR) operation was performed, the rmsd of ESP was slightly larger, but the molecular dipole moment was almost the same. The induced dipoles of the molecular block occur in proportion to the point charges of the adjacent molecular blocks. For this reason, it is extremely important to reproduce the charge distribution of molecular blocks as accurately as possible and to optimize the point charge so that the total charge is also an integer.
Even when the number of points on the cytosine surface is reduced by half (169 points), the rms deviation of ESP tends to be almost the same as shown in the table below. Here the charges of the two hydrogens (H41 and H42) of the amino group are optimized to the same value. The molecular dipole moment is the same as the 337 points optimization. In the case of 155 points where capped hydrogen removal (CHR) operation was performed, the rmsd of ESP was slightly larger, but the molecular dipole moment was almost the same. The induced dipoles of the molecular block occur in proportion to the point charges of the adjacent molecular blocks. For this reason, it is extremely important to reproduce the charge distribution of molecular blocks as accurately as possible and to optimize the point charge so that the total charge is also an integer.
| Atom | Kosugi 1.8 / 337 points | Half 169 points | CHR 155 points |
|---|---|---|---|
| H1 | 0.369 | 0.367 | - |
| N1 | -0.707 | -0.708 | 0.254 |
| C6 | 0.361 | 0.370 | -0.267 |
| H6 | 0.120 | 0.118 | 0.263 |
| C5 | -0.883 | -0.890 | -0.589 |
| H5 | 0.263 | 0.263 | 0.226 |
| C4 | 1.218 | 1.216 | 0.899 |
| N4 | -1.168 | -1.170 | -1.015 |
| H41 | 0.478 | 0.473 | 0.436 |
| H42 | 0.466 | 0.473 | 0.436 |
| N3 | -0.899 | -0.912 | -0.693 |
| C2 | 1.027 | 1.040 | 0.545 |
| O2 | -0.643 | -0.645 | -0.524 |
| ESP RRMSD | 3.56 % | 3.54 % | 4.73 % |
| μ Tot (bebye) | 6.67 | 6.67 | 6.73 |