D. Polarized One-electron Potential Fitting
Atom Dipole Interaction Model
In induced dipole force field, the polarization energy (Eplz) is expressed as follows
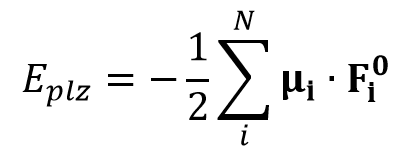
Here, μi is the induced dipole moment at atom i and Fi0 is the applied electrostatic field at atom i. The induced dipole moments are calculated as follows
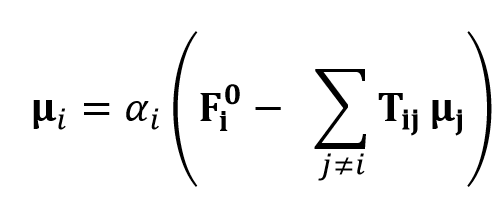
Here, αi is thr polarizabilitiy of atom i and μj is the induced dipole moment of the atom other than i. Tij is the dipole field tensor. Several damping types have been introduced for repairing the deficiency of infinit polarization.
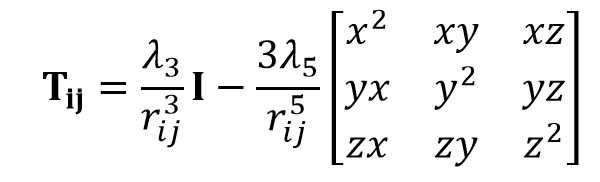
where I is the unit matrix, rij is the distance between atoms i and j. λ3 and λ5 are the dumping functions. Damping types of Simple scaling (S), Exponential (E), Linear (L), and Gaussian (G) have been used.5
POP Optimization Polarized one-electron potential (POP) optimization is a method of determining atomic polarizabilities. 2-5 In this method, the amount of change in ESPs mapped on the molecular surface induced by an external test charge are first estimated using the QM method. There, a series of potential maps are needed that vary with the test charge placed on the molecular surface. The molecular surface defined by an envelope of 1.8 times the van der Waals radius of the atoms and a test charges of +0.1 e are used. Next, the atomic polarizabilities (αi) are optimized to reproduce the surface potentials derived from the QM calculations. The POP optimization is similar to ESP optimization for determining the atomic charges of molecules. While ESP optimization targets the ESPs on a molecular surface, the POP optimization targests the change of ESPs on molecular surface.
In POP optimization Fi0 is very simple as follows
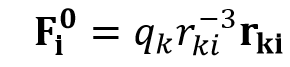
Here, a procedure for POP optimization is described.
1. Prepare two files required for POP optimization for dumping type S3
"r3_01_C" file prepares the data required for POP optimization. The dumping type is a simple scaling of λ3 and λ5 with 0.3 (S3). The meaning of each line is as follows. The data for cytosine, taken here as an example, are shown in parentheses.
Download
"lin_s03111_C.dat" file prepares mainly the dumping data. The meaning of each line is as follows. The data for cytosine are shown in parentheses.
Download
Here, μi is the induced dipole moment at atom i and Fi0 is the applied electrostatic field at atom i. The induced dipole moments are calculated as follows
Here, αi is thr polarizabilitiy of atom i and μj is the induced dipole moment of the atom other than i. Tij is the dipole field tensor. Several damping types have been introduced for repairing the deficiency of infinit polarization.
where I is the unit matrix, rij is the distance between atoms i and j. λ3 and λ5 are the dumping functions. Damping types of Simple scaling (S), Exponential (E), Linear (L), and Gaussian (G) have been used.5
POP Optimization Polarized one-electron potential (POP) optimization is a method of determining atomic polarizabilities. 2-5 In this method, the amount of change in ESPs mapped on the molecular surface induced by an external test charge are first estimated using the QM method. There, a series of potential maps are needed that vary with the test charge placed on the molecular surface. The molecular surface defined by an envelope of 1.8 times the van der Waals radius of the atoms and a test charges of +0.1 e are used. Next, the atomic polarizabilities (αi) are optimized to reproduce the surface potentials derived from the QM calculations. The POP optimization is similar to ESP optimization for determining the atomic charges of molecules. While ESP optimization targets the ESPs on a molecular surface, the POP optimization targests the change of ESPs on molecular surface.
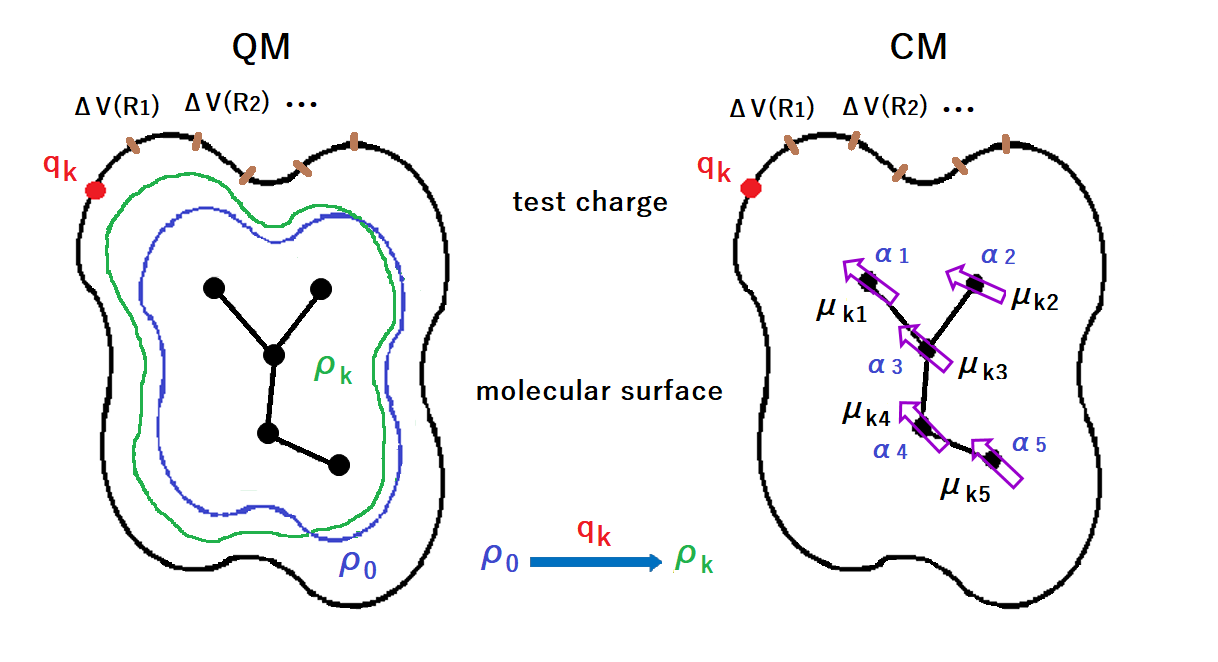
Figure D1. Schematic diagram of POP optimization.
QM stands for quantum mechanics and CM for classical mechanics.
ρ0 is the electron density of molecule not affected by the test charge qk,
and ρk is the electron density of molecule under the influence of the test charge qk.
The qk is placed on the molecular surface.
ΔV is the amount of change in electrostatic potential at the molecular surface for ρ0 and ρk.
In CM, the electron density change by the test charge qk is represented by induced dipole moments (μki).
The arrows indicate the direction of electron movement.
αi is the atomic polarizabilitiy.
The polarizabilities are optimized in order to reproduce the surface electrostatic potensial differences.
In POP optimization Fi0 is very simple as follows
Here, a procedure for POP optimization is described.
- Prepare two files required for POP optimization for dumping type S3
- Perform POP optimization and determine the atomic polarizabilities using the POP file prepared in G09
- Prepare two files required for POP optimization for dumping type G
- Reproducibility of molecular polarizability
1. Prepare two files required for POP optimization for dumping type S3
"r3_01_C" file prepares the data required for POP optimization. The dumping type is a simple scaling of λ3 and λ5 with 0.3 (S3). The meaning of each line is as follows. The data for cytosine, taken here as an example, are shown in parentheses.
1) Number of molecular surface shell (1: one shell); type of fitting (0: atomic polarizability); irest dumping type (3: simple scaling).
2) Magunitude of a test charge (0.1: +0.1e)
3) Type of polarizabilitiy (4: atom).
4) Number of bond polarizabilities (0);Number of atom polarizabilities (13).
5) The next 13 lines define atom number and the initial value of polarizability (au).
("1 1.0" ... "13 8.0":initial value of atom 1 is 1.0 au ... initial value of atom 13 is 8.0 au).
If the initial values are exactly the same number, the values are treated as one variable.
Atomic numbers are assigned as initial values here.
6) The next line is the number of test charge used (19).
7) The next 19 lines are the x,y,z coordinate of test charges. Those are taken from "extfldxyz.dat" in "G09 Test Chg" page.
2) Magunitude of a test charge (0.1: +0.1e)
3) Type of polarizabilitiy (4: atom).
4) Number of bond polarizabilities (0);Number of atom polarizabilities (13).
5) The next 13 lines define atom number and the initial value of polarizability (au).
("1 1.0" ... "13 8.0":initial value of atom 1 is 1.0 au ... initial value of atom 13 is 8.0 au).
If the initial values are exactly the same number, the values are treated as one variable.
Atomic numbers are assigned as initial values here.
6) The next line is the number of test charge used (19).
7) The next 19 lines are the x,y,z coordinate of test charges. Those are taken from "extfldxyz.dat" in "G09 Test Chg" page.
Download
"lin_s03111_C.dat" file prepares mainly the dumping data. The meaning of each line is as follows. The data for cytosine are shown in parentheses.
1) Just a title for the next line. It indicates 1-5 or more interactions.
2) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0);
cut off distance for charge-induced dipole (999.0: no cut off);cut off distance for induced dipole-dipole (999.0: no cut off);
dumping factor for E1, E2, L, and G type (0.0: not used for S type).
3) Just a title for the next line. It indicates 1-2 interactions.
4) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (0.3: S3 type);
5) The next 13 lines define bond (i-j)(format: 200i4). Atom number i (1);the number j of the partner atom forming the bond (2).
j is greater than i to avoid double counting (i<j).
6) Just a title for the next line. It indicates 1-3 interactions.
7) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: S3 type).
8) Just a title for the next line. It indicates 1-4 interactions.
9) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: S3 type).
10) Just a title for the next line. It indicates that the next data are atomic charges.
11) The next 15 lines define atom number and the atomic charge (blank: not used).
2) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0);
cut off distance for charge-induced dipole (999.0: no cut off);cut off distance for induced dipole-dipole (999.0: no cut off);
dumping factor for E1, E2, L, and G type (0.0: not used for S type).
3) Just a title for the next line. It indicates 1-2 interactions.
4) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (0.3: S3 type);
5) The next 13 lines define bond (i-j)(format: 200i4). Atom number i (1);the number j of the partner atom forming the bond (2).
j is greater than i to avoid double counting (i<j).
6) Just a title for the next line. It indicates 1-3 interactions.
7) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: S3 type).
8) Just a title for the next line. It indicates 1-4 interactions.
9) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: S3 type).
10) Just a title for the next line. It indicates that the next data are atomic charges.
11) The next 15 lines define atom number and the atomic charge (blank: not used).
Download
2. Perform POP optimization and determine the atomic polarizabilities using the POP file prepared in G09
Download
"pop_s03111_01_C.out" is a output file. The optimized atomic polarizabilities are obtained.
Download
"fin_plz_s03111_01_C.dat" is the optimized polarizability extraction file.
Download
"POPopt_mi_2023.f" is a Fortran program for polarizability optimization using POP. A nonlinear optimization (Levenberg-Marquardt) procedure is used. If you downloaded and used the POPopt_mi_2023 program, please inform S. Nakagawa that you used it (naka(a)kinjo-u.ac.jp). Also, let me know if you find any bugs.
Download
Householder method is used for the matrix inversion. The code for Fortran 90 version by J.-P. Moreau is taken from Web site (http:/www.jpmoreau.fr). (http://jean-pierre.moreau.pagesperso-orange.fr/Fortran/householder_f90.txt) (Ref. "Methodes de calcul numerique - Tome 2 - By Claude, Nowakowski, PSI Editions, 1984")
"POPopt_mi.cm" is a sample csh command to run POP optimization.
1) "rest_2_3_3_C" (rest_2) is the input POP file (see "G09_Tst_Chg" page).
2) "r3_01_C" (rest_3) is the input file above.
3) "lin_s03111_C.dat" is the input file above.
4) "pop_s03111_01_C.out" is the output file.
5) The optimized atomic polarizabilities (au) are written in "fin_plz_s03111_01_C.dat".
2) "r3_01_C" (rest_3) is the input file above.
3) "lin_s03111_C.dat" is the input file above.
4) "pop_s03111_01_C.out" is the output file.
5) The optimized atomic polarizabilities (au) are written in "fin_plz_s03111_01_C.dat".
Download
"pop_s03111_01_C.out" is a output file. The optimized atomic polarizabilities are obtained.
Download
"fin_plz_s03111_01_C.dat" is the optimized polarizability extraction file.
The data of capped hydrogen (first line) was deleted manually.
Download
"POPopt_mi_2023.f" is a Fortran program for polarizability optimization using POP. A nonlinear optimization (Levenberg-Marquardt) procedure is used. If you downloaded and used the POPopt_mi_2023 program, please inform S. Nakagawa that you used it (naka(a)kinjo-u.ac.jp). Also, let me know if you find any bugs.
Download
Householder method is used for the matrix inversion. The code for Fortran 90 version by J.-P. Moreau is taken from Web site (http:/www.jpmoreau.fr). (http://jean-pierre.moreau.pagesperso-orange.fr/Fortran/householder_f90.txt) (Ref. "Methodes de calcul numerique - Tome 2 - By Claude, Nowakowski, PSI Editions, 1984")
3. Prepare two files required for POP optimization for dumping type G
"r3G_01_C" file prepares the data required for POP optimization. The dumping type is Gaussian (G) with a dumping factor 0.926. The meaning of each line is as follows. The data for cytosine, taken here as an example, are shown in parentheses.
Download
"lin_G0926_C.dat" file prepares mainly the dumping data. The meaning of each line is as follows. The data for cytosine are shown in parentheses.
Download
Executing the csh command POPopt_mi.cm according to Section 2 yields the following output. "pop_G0926_01_C.out" is a output file. G-type optimized atomic polarizabilities can be obtained.
Download
"r3G_01_C" file prepares the data required for POP optimization. The dumping type is Gaussian (G) with a dumping factor 0.926. The meaning of each line is as follows. The data for cytosine, taken here as an example, are shown in parentheses.
1) Number of molecular surface shell (1: one shell); type of fitting (0: atomic polarizability); irest dumping type (6: Gaussian).
2) Magunitude of a test charge (0.1: +0.1e)
3) Type of polarizabilitiy (4: atom).
4) Number of bond polarizabilities (0);Number of atom polarizabilities (13).
5) The next 13 lines define atom number and the initial value of polarizability (au).
("1 1.0" ... "13 8.0":initial value of atom 1 is 1.0 au ... initial value of atom 13 is 8.0 au).
If the initial values are exactly the same number, the values are treated as one variable.
Atomic numbers are assigned as initial values here.
6) The next line is the number of test charge used (19).
7) The next 19 lines are the x,y,z coordinate of test charges. Those are taken from "extfldxyz.dat" in "G09 Test Chg" page.
2) Magunitude of a test charge (0.1: +0.1e)
3) Type of polarizabilitiy (4: atom).
4) Number of bond polarizabilities (0);Number of atom polarizabilities (13).
5) The next 13 lines define atom number and the initial value of polarizability (au).
("1 1.0" ... "13 8.0":initial value of atom 1 is 1.0 au ... initial value of atom 13 is 8.0 au).
If the initial values are exactly the same number, the values are treated as one variable.
Atomic numbers are assigned as initial values here.
6) The next line is the number of test charge used (19).
7) The next 19 lines are the x,y,z coordinate of test charges. Those are taken from "extfldxyz.dat" in "G09 Test Chg" page.
Download
"lin_G0926_C.dat" file prepares mainly the dumping data. The meaning of each line is as follows. The data for cytosine are shown in parentheses.
1) Just a title for the next line. It indicates 1-5 or more interactions.
2) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: this value is not used for G type);
cut off distance for charge-induced dipole (999.0: no cut off);cut off distance for induced dipole-dipole (999.0: no cut off);
dumping factor for E1, E2, L, and G-type (0.926: G-type).
3) Just a title for the next line. It indicates 1-2 interactions.
4) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: this value is not used for G type);
5) The next 13 lines define bond (i-j)(format: 200i4). Atom number i (1);the number j of the partner atom forming the bond (2).
j is greater than i to avoid double counting (i<j).
6) Just a title for the next line. It indicates 1-3 interactions.
7) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: this value is not used for G type).
8) Just a title for the next line. It indicates 1-4 interactions.
9) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: this value is not used for G type).
10) Just a title for the next line. It indicates that the next data are atomic charges.
11) The next 15 lines define atom number and the atomic charge (blank: not used).
2) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: this value is not used for G type);
cut off distance for charge-induced dipole (999.0: no cut off);cut off distance for induced dipole-dipole (999.0: no cut off);
dumping factor for E1, E2, L, and G-type (0.926: G-type).
3) Just a title for the next line. It indicates 1-2 interactions.
4) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: this value is not used for G type);
5) The next 13 lines define bond (i-j)(format: 200i4). Atom number i (1);the number j of the partner atom forming the bond (2).
j is greater than i to avoid double counting (i<j).
6) Just a title for the next line. It indicates 1-3 interactions.
7) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: this value is not used for G type).
8) Just a title for the next line. It indicates 1-4 interactions.
9) Dumping factor for charge-induced dipole interaction (0.0); dumping factor for induced dipole-diple (1.0: this value is not used for G type).
10) Just a title for the next line. It indicates that the next data are atomic charges.
11) The next 15 lines define atom number and the atomic charge (blank: not used).
Download
Executing the csh command POPopt_mi.cm according to Section 2 yields the following output. "pop_G0926_01_C.out" is a output file. G-type optimized atomic polarizabilities can be obtained.
Download
4. Reproducibility of molecular polarizability
The POP optimization results (pop_s03111_01_C.out and pop_G0926_01_C.out) of cytosine are summarized in Table below. The polarization parameters obtained from QM calculations using diffuse functions are known to lead to over-polarization of the condensed system. The atomic polarizabilities were mainly optimized with reference to the POPs calculated using wB97XD/cc-pVTZ, which lacks diffuse functions. The exact molecular polarizability can be obtained by G09 calculation with "polar" keyword. The isotropic molecular polarizability of wB97XD/cc-pVTZ is reproduced well the experimental value. The molecular polarizabilities of Simple scaling (S3) and Gaussian (G) types reproduced well the QM results.
Unit is Bohr3. Unit of RMSD is kcal/mol.
The POP optimization results (pop_s03111_01_C.out and pop_G0926_01_C.out) of cytosine are summarized in Table below. The polarization parameters obtained from QM calculations using diffuse functions are known to lead to over-polarization of the condensed system. The atomic polarizabilities were mainly optimized with reference to the POPs calculated using wB97XD/cc-pVTZ, which lacks diffuse functions. The exact molecular polarizability can be obtained by G09 calculation with "polar" keyword. The isotropic molecular polarizability of wB97XD/cc-pVTZ is reproduced well the experimental value. The molecular polarizabilities of Simple scaling (S3) and Gaussian (G) types reproduced well the QM results.
| method | wB97XD/cc-pVTZ | S3 | G 0.926 | Exptl |
|---|---|---|---|---|
| αH | - | 1.20 | 1.32 | - |
| αC | - | 7.27 | 11.10 | - |
| αN | - | 5.33 | 7.54 | - |
| αO | - | 4.28 | 5.20 | - |
| POP opt RMSD (RRMSD) | 0.0 | 0.053 (7.1 %) | 0.049 (6.5 %) | - |
| αmolxx | 95.16 | 96.15 | 93.45 | - |
| αmolyy | 72.85 | 74.87 | 76.44 | - |
| αmolzz | 36.40 | 35.79 | 36.69 | - |
| αmolisotropic | 68.13 | 68.94 | 68.86 | 69.5 |